ATAC-Seq
Overview
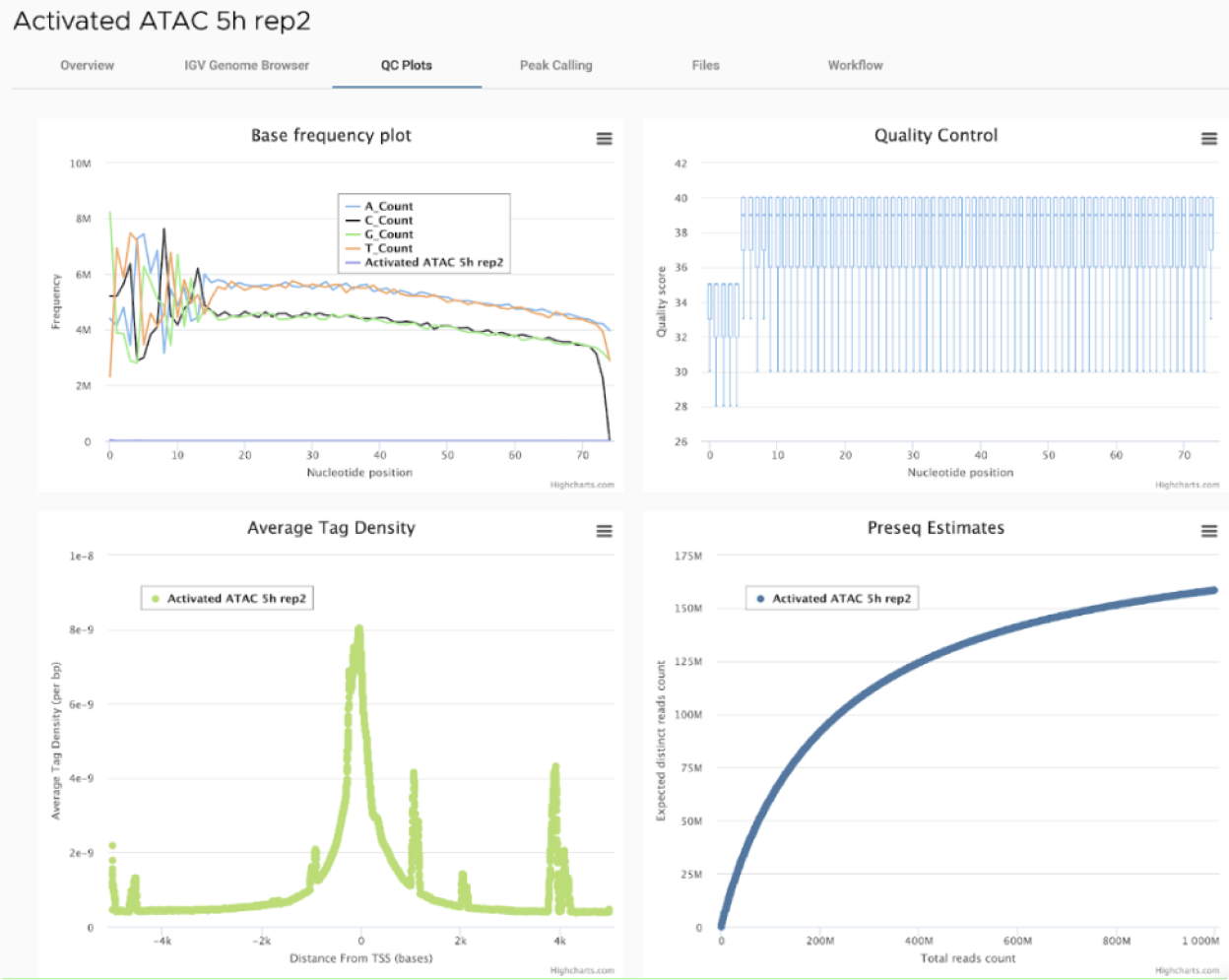
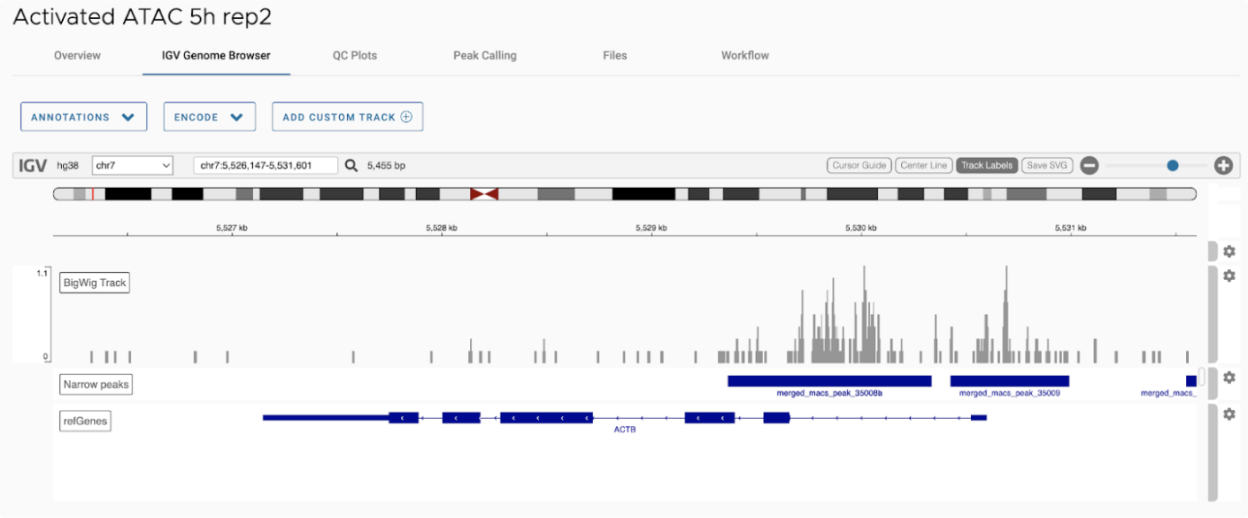
ATAC-Seq is a next-generation sequencing (NGS) application that identifies open chromatin regions. Open chromatin refers to areas of DNA that are not protected by nucleosomes or other proteins, and often accompany sites of transcription factor binding, enhancers, and active promoters and are associated with gene activation. ATAC-Seq improves the sensitivity and input requirements of historical methods for identifying open chromatin including DNAseI Hypersensitivy Assays and FAIRE-Seq, and can be used with single cells methods. Omni-ATAC is a modified ATAC procedure that reduces mitochondrial background reads and provides consistent results with 50,000 cells.
General Workflow
ATAC-Seq utilizes a Tn5 transposase enzyme to insert sequencing adapters directly into accessible areas of double stranded DNA. Cells are first washed, then permeabilized with digitonin to allow for transposase diffusion. During incubation with Tn5, NGS platform adapters are tagged to DNA within accessible regions. The DNA is purified, and PCR extension and amplification is performed to create an NGS library.
Data Analysis
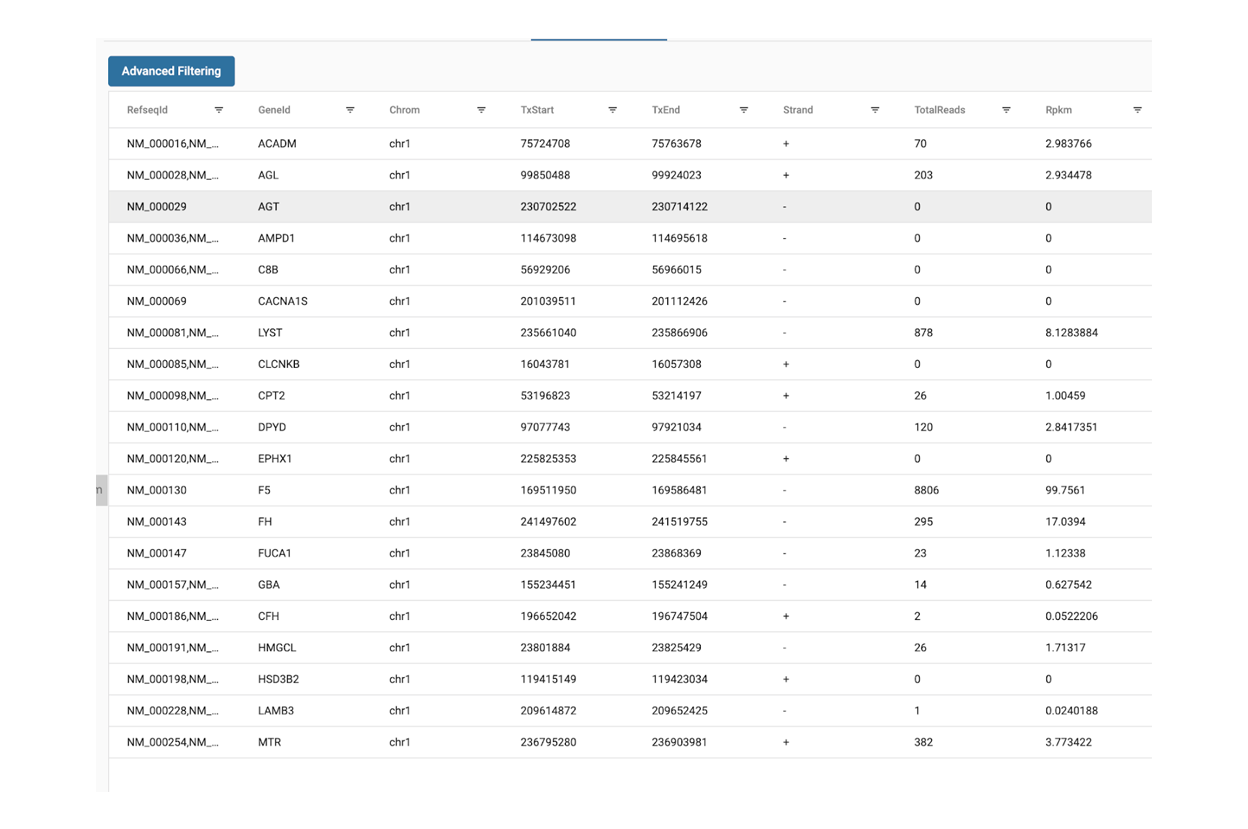
SciDAP is a no-code bioinformatics platform that enables biologists to analyze NGS-based data without a bioinformatician. It has built-in pipelines based on open-source workflows to analyze bulk data from ATAC-Seq libraries. SciDAP starts from the FASTQ files provided by most DNA core facilities and commercial service providers. Starting from raw data allows SciDAP to ensure that all experiments have been processed in the same way and simplifies the deposition of data to GEO upon publication. The data can be uploaded from the end-users’ computer, downloaded directly from an FTP server of the core facility by providing a URL, or from GEO by providing SRA accession number.